
While we enjoyed a smooth, full-term pregnancy and delivery, Cleo was unusually subdued as a newborn. She had an ashen complexion despite repeated normal bilirubin levels, she never cried, and would only feed briefly before falling back asleep. Cleo is my third child. My first two were premature (twins). They were far more alert than Cleo, despite her being almost twice their size at birth. I just sensed something was very wrong. The nurses reassured me that infants are often sluggish at first and to make the most of the peace and quiet as it would be short lived. The lethargy persisted, however, and she never woke up crying for a feed, so I would set an alarm every 3 hours at night to ensure she was fed often enough.
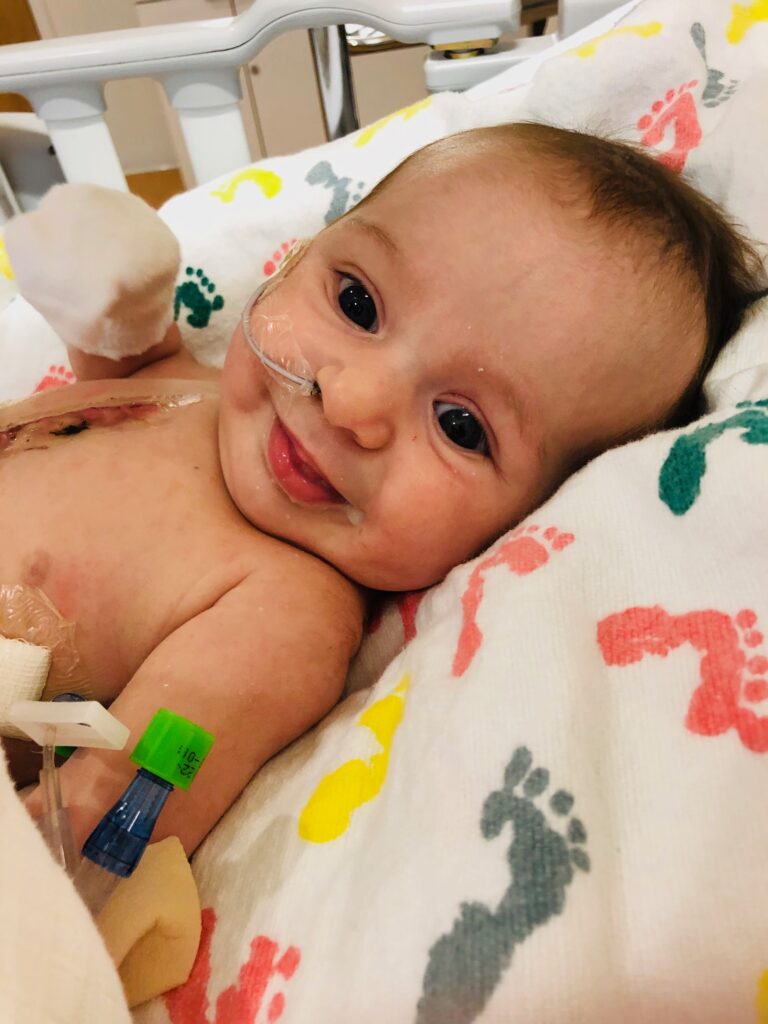
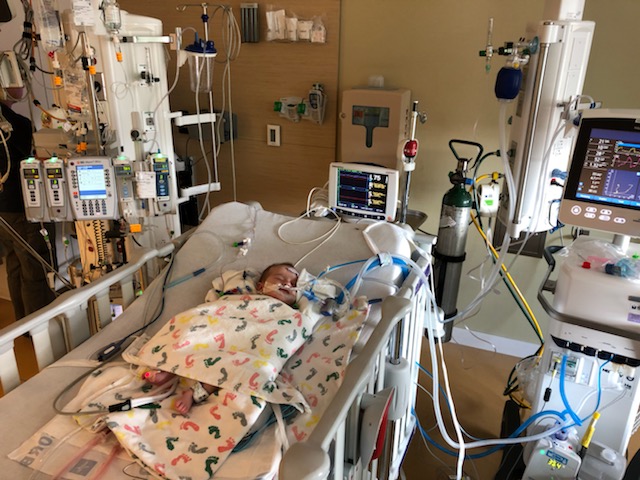
Before we were discharged from the hospital, a pediatrician detected a heart murmur and referred Cleo to a cardiologist. Further testing found a sizable ASD and VSD (a hole between each of the upper and lower chambers of her heart). Initially, everyone naturally blamed Cleo’s reluctance to feed on that. The only person who felt otherwise was Cleo’s cardiologist who was adamant this was not yet cardiac related. In a sense the heart issues served as a red herring. At 2 weeks old, Cleo began to vomit frequently after feeding. She was born at the 75th percentile for weight but by 10 weeks had dropped to 1st percentile. At this point, Cleo was admitted to hospital for failure to thrive and an NG tube was placed. She was also showing some signs of early heart failure so was started on a diuretic. While the diuretic seemed to help, the vomiting continued and the various specialists who saw her found no other issues to explain her failure to thrive. After a week in hospital, the team decided to proceed with open heart surgery to patch the holes. It was a long surgery (6 hours) but everything went well and besides some initial high blood pressures, she sailed through without complication. We all hoped her feeding would pick up thereafter but at the back of my mind I was skeptical. While her ashen appearance transformed into a pink healthy glow as she recovered in the PICU, her appetite remained poor and the vomiting continued. I was instructed to fortify my breastmilk with formula to boost her caloric intake but that only made matters worse. It was as if she could not tolerate her food but all tests were fruitless. She was perfectly happy until we switched the feeding machine on, to which she’d become visibly uncomfortable. The cardiology team was stumped and the feeding team told us that sometimes we just don’t know why a baby won’t feed. Being told there may just be no answer was devastating.

At 5 months old, we reluctantly agreed for the NG to be replaced with a g-tube after she repeatedly regurgitated the tube while vomiting. We accepted that this was just part of her and our lives for the time being. In the meantime, we involved Growing Independent Eaters, a private tube-weaning consultancy, with the hope of weaning her from tube feeding. By 9 months old, her feeding did actually start to improve after we reverted back to expressed milk only and reduced her tube feed volume. We finally felt optimistic; maybe time was all she needed? She also finally started taking some solids. While she was somewhat delayed, she was now able to sit up by herself and was making good progress toward rolling and crawling. Then, at 9.5 months she became ill with a cold. While her cold symptoms abated, during the course of two weeks, she became increasingly fussy and the only thing that comforted her was to be held and rocked back and forth day and night. This was in contrast to a very happy child who barely ever cried. The fussiness reached what felt like a crescendo when she suddenly fell unresponsive for 5 minutes and then started to seize. Altogether the episode lasted about 10 minutes by which point the EMS team was there. Imaging and an initial 30 minute EEG found nothing amiss. We were told it was just a febrile seizure but, as it so happened, I had recorded a normal temperature just minutes prior. We saw a neurologist and I immediately mentioned her fussiness (which continued post-seizure and was very apparent during the appointment) but she had no explanation.
A week later she had her next seizure and was then started on epilepsy medication. An MRI found diffuse cerebral microhemorrhage but why we did not know. The neurologist again wondered if it was a byproduct of her heart surgery and time spent on the heart-lung bypass machine. Each seizure was associated with a temporary loss of skills. She lost the ability to sit up, her hand movements were uncoordinated, she showed no interest in play, and was too fussy to eat so we sadly reverted back to tube feeding. The only thing that brought her comfort was being held for hours on end.
After starting her first anti-epileptic medication, her fussiness subsided somewhat but she still wasn’t her old self. She’d make sudden jerking movements as if she’d been given an electric shock and her head would frequently bob up and down while she zoned out for a few seconds at a time before coming back. Full blown unresponsive seizures continued and gradually became more frequent, clustered, and prolonged. She went into status 7 times. We were given rescue medication to administer but it was ineffective, and some days had to use it more than once. Her meds were increased several times and a 2nd medication was added. We became accustomed to dropping everything in order to rush her to hospital but once we were there, very little was done besides monitoring and increasing epilepsy meds that were clearly not working for her. She had a growing collection of cuddly toys from the many ambulance trips and children’s hospital visits.
Each time we saw the neurologist, I left feeling like she played down the severity of the situation. One time she informed us that she’d had another patient have a seizure lasting hours who had come out of it “just fine”. I think she was trying to reassure us but in reality it did the very opposite. She also seemed unconvinced by the many brief seizures we were observing, sometimes scores of myotonic jerks during the course of the day. We were told they were developmentally normal even though they only started happening after the first seizure. I happened across a genetic epilepsy panel that Cleo was eligible for (thanks to a helpful parent in a FB group for parents of children with epilepsy). When I asked about it, however, the neurologist was dismissive and said “cases like Cleo’s are almost never genetic”. The prolonged nature of the seizures, however, were unusual (most seizures are well under 5 minutes long), her seizures seemed to be mixed in type, which again is less common, and her fussiness and sleeplessness were striking (epilepsy meds usually cause increased sleepiness not the opposite). My thought was, what harm could it do to at least run the panel? After asking several times, begging even, she finally agreed. 3 months of seizures had passed by the time the test was ordered. In the interim, we had pushed for a second opinion but the waiting time was months so we were in a stalemate with the current neurologist. The results identified 2 variants involving the antiquitin gene, both associated with PDE. I immediately looked it up. “PDE is often characterized by prolonged seizures with associated fussiness”. I knew we very likely had our answer even though it was an atypical presentation given the late seizure onset. I felt a sense of relief and hope. Diagnostic limbo is a very hard place to be, especially when your gut is telling you that something is very wrong. A urine test was ordered for alpha-AASA (we were not informed how to store the urine, i.e. to freeze it immediately, so it’s a small miracle that the sample even gave a positive result) to confirm the suspected diagnosis. As soon as pyridoxine was started, the seizures thankfully stopped (including the many brief episodes we’d observed that had been categorically dismissed by her medical team) and our beautiful, happy girl re-emerged from the fog. The first 24 hours on pyridoxine, however, were not smooth. Cleo became extremely disoriented as if intoxicated, could barely hold her head up, vomited repeatedly, and then sank into a deep sleep for 15 hours that was difficult to rouse her from (I did not sleep that night and called the neurology team who advised us to monitor her at home rather than bringing her in). At the time, we assumed it was unrelated as pyridoxine is just a benign vitamin (B6) and she’d recently started another anti-epileptic drug (AED) but I later read that starting this medication can cause transient cerebral suppression in infants with PDE and should be undertaken in an ICU setting. In retrospect, I wish she had been hospitalized and monitored for her safety but one of the pitfalls of a rare disorder is that often the medical team knows so little that they are not equipped to give adequate guidance. We initially had to figure out what dose of B6 to give our daughter based on the medical literature, for example. Fortunately, we’re now with a team in the neighboring state that is far more knowledgeable about PDE and corresponds closely with the research team in Colorado.


We have successfully weaned Cleo off of the AEDs and she has not had another seizure since starting treatment. Once we were under the care of a metabolic geneticist, Cleo began arginine therapy, a strict low protein diet and medical formula. She was feeding orally by this point but refused the formula despite trying all the tricks so we had to feed her it via her g-tube. As soon as we did so, however, her appetite dwindled and after 3 weeks she’d lost nearly 10% of her body weight. This obviously wasn’t sustainable so we made the decision to stop the medical formula and to moderately restrict her protein (18g protein / day). Switching a 14 months old to a completely different diet was tough and a steep learning curve. We had to educate ourselves on protein content overnight and with little support. The local team provided very few resources unfortunately (we are now lucky to have a team with a far more educational and hands-on approach and it has made a world of difference to us as a family). Cleo is growing beautifully and taking all solids and liquids by mouth. She was finally ready to have her g-tube removed when she was 2.5. It was a big decision as it was such a convenient way of delivering her daily meds and a means of getting nutrition into her in times of illness, and there have been times where we have had to go to the ER to get IV meds and fluids where had we kept the tube, we might have been able to manage the illness at home.
While Cleo was delayed as an infant, early intervention, speech and physical therapy in the early years have helped bring her back on track, and as a preschooler she’s meeting all of her developmental milestones, thriving in school and no longer receives any therapies. We did have her attend a Montessori school last year where independence and self-direction were highly encouraged, and in that setting she did run into some difficulties with remembering the various steps she needed to take. This would result in freezing episodes where she would stand motionless for a minute or two at a time before someone intervened to help her. In her current school, however, she’s had no such issues and her teachers have told us they’d never guess what she’d been through previously.
She’s 4.5 now, very social and chatty, and has a rich imagination; she loves to play doctors and her poor dolls receive a lot of shots and are covered in bandages! She’s also a natural climber and does very well keeping up with her big twin brothers. We are so excited for her future but at the same time wonder what effect the diagnostic delay, the seizures and all the challenges she faced in her first year will have on her long-term. We even wonder whether she could have avoided open heart surgery had she been diagnosed and treated for PDE soon after birth. Her story is yet to be written of course but I think there will always be a question in my mind of “how would things be different had we known about PDE at birth?”. At the very least, I imagine we could have avoided the feeding tube and had a much more normal first year of life. Raising twins was hard but raising a child with a rare, undiagnosed metabolic disorder was a whole new level of hard and quite isolating, especially in the midst of the pandemic. Watching her go through open heart surgery was nothing compared to helplessly witnessing her in a state of metabolic encephalopathy without knowing what was happening to our precious baby. Children with a later onset of seizures tend to fare better than those with classic PDE but the length of diagnostic delay is the biggest determiner of developmental outcome. There is so much that is still unknown about this condition, such as why some children start to seize earlier than others or why some do better than others (Cleo’s antiquitin enzyme is completely lacking yet she has fortunately had a good outcome so far, developmentally, for example). For that, we are so grateful. Her extremely happy and social demeanor despite what she’s been through reminds us daily that we are very fortunate and honored to have Cleo as our daughter. She is such a happy and resilient little girl!
We are so grateful to CurePDE and the various research teams around the world (especially Dr. Coughlin and his team in Colorado) for all the work they are doing. We were so fortunate to attend the first family weekend in Texas. It was surreal finally meeting other families and their beautiful children with PDE and sharing our common experiences of all the trials we endured to find a diagnosis and access the best treatment to allow our children to thrive in spite of PDE. I had no idea how close we might be to newborn screening of PDE, nor what a labor of love it has taken to reach this point, and even a possible curative treatment in the not so distant future. Cleo loves protein, much like most kids crave candy. She doesn’t care about candy but desires anything high in protein, it’s uncanny and challenging. The idea that one day she can eat whatever she wants without us always having to keep check is something we are so excited about. We are also excited for future families of children with PDE not having to go through what we did with Cleo. A diagnosis of a rare metabolic disorder is a shock but imagine a time when you find out within a week of your child’s birth and have an effective treatment that will protect them from seizures and developmental delay, and allow them to follow a normal diet. I truly believe CurePDE has come at the right time to provide the momentum toward this goal.

