
We wrote this story about Andre in 2014 when he was 3 years old. We wanted to post it as it was to be able to reflect back on where we were and where we are now.
Our Journey with Pyridoxine-Dependent Epilepsy (PDE)
Our story began on a summer day, July 17, 2011, at Cedars-Sinai Medical Center in Beverly
Hills, CA. My pregnancy had been uneventful, and with my second child on the way, I was filled
with confidence and anticipation. My first child, Jenny, was a healthy teenager at that time, and I
had no reason to suspect that anything would be different with Andre.
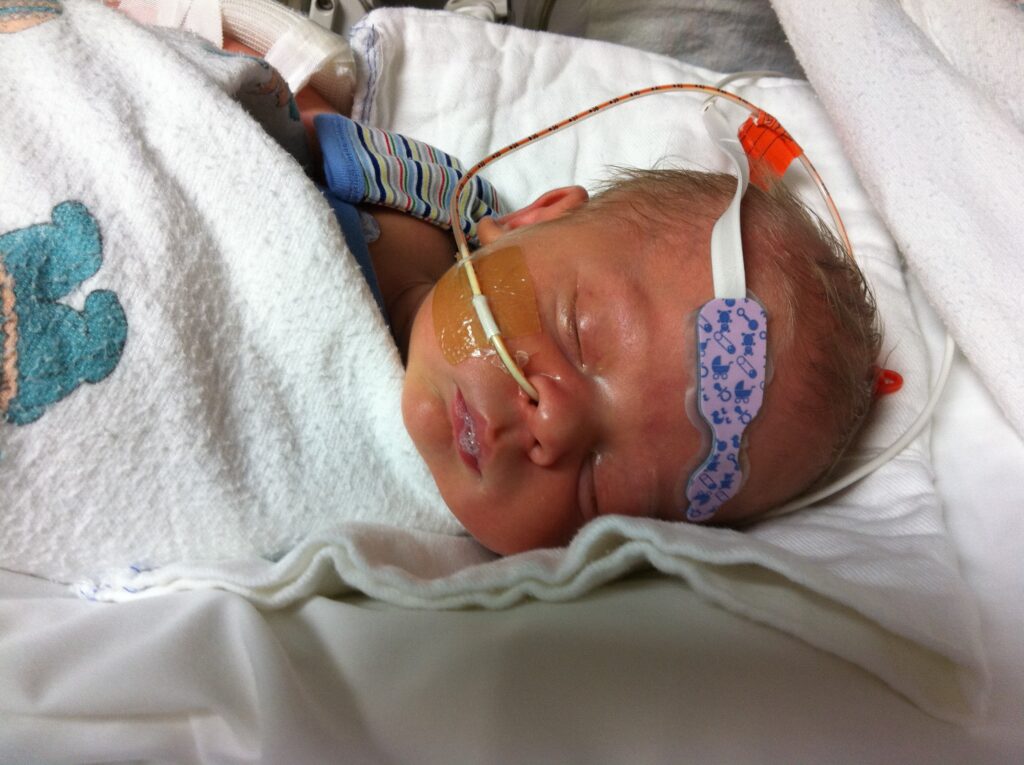
The delivery went smoothly, but there was a subtle difference that raised concerns—Andre was
noticeably jittery upon his arrival, catching the attention of the medical team. In response, they
conducted a blood sugar test, which fortunately returned within normal limits. However, his
behavior didn’t align with our prior experience with Jenny. Instead of peaceful sleep, he barely
slept and, rather than the typical cries, emitted short, piercing screams throughout the night.
Despite these early signs, we tried to reassure ourselves that everything was alright, but a
mother’s intuition can be powerful, and deep down, we knew something needed attention.
That morning, something peculiar began to unfold. Andre refused to breastfeed and, after being
fed with a milk formula, he vomited. The doctors did not seem to be concerned and discharged
us to go home. While I was walking out of the room, holding Andre in my arms, I looked at him
and saw that his skin took on an alarming blue hue and he seemed to have stopped breathing.
In panic, I screamed for a nurse. A nurse rushed into the room and took Andre immediately.
Several people ran into the nursery. I heard someone say they were able to bring him back.
I walked into the nursery and saw under a bright heat lamp. His tiny hands and legs were
shaking.
I was told he needed to be transferred to the NICU and shortly after they rolled him into the
elevator and into the NICU.
Andre continued to shake, and scream, leaving doctors bewildered. The seizures continued with
some intervals as Andre was not able to feed and was put on a feeding tube. The doctors were
indecisive and said they had never seen anything similar before.
The medical staff at Cedars-Sinai, renowned for their expertise, puzzled over Andre’s condition.
More than 30 neurology specialists were consulted, yet none could identify the cause. Andre
continued to have prolonged episodes of convulsions, sometimes lasting overnight and doctors
worked hard trying to stabilize him.
Andre’s primary neurologist initially prescribed Keppra (Levetiracetam), an anticonvulsant used
to treat epilepsy. However, it had no effect, and we were assured that it needed time to work. It
became apparent that continuing down this path would yield no answers.
Their uncertainty was disheartening. Numerous tests were conducted along with MRI. At some
point our neurologist called us in and informed us that MRI showed that Andre incurred hypoxic
ischemic injury and half of his brain was shrunk. She informed us that Andre will never walk or
talk and will continue to deteriorate.
The only consensus was that this was not classical epilepsy, as Andre’s electroencephalogram
showed no detectable abnormalities (a conclusion we later came to doubt).
After a month we were sent home with a diagnosis of a rare condition that did not really match
our symptoms while the results of the genetic testing were pending.
After coming home, all the symptoms returned. Andre continued to vomit his food, screamed,
his arms and legs were jerking and was unable to sleep sometimes for 36 hours. We were in
panic and despair.
We made a decision to go to a different hospital in search of answers. Upon arrival to UCLA
Hospital’s emergency room we were assigned to Dr. Jason Lerner. Dr Lerner immediately put
Andre on EEG and monitored his brain activity. After two hours of convulsing Andre’s EEG
showed a myoclonic seizure. Dr. Lerner recognized the epileptic nature of Andre’s seizures,
contradicting our earlier diagnosis. A single injection of phenobarbital brought relief within
minutes, and B6-dependent epilepsy was suspected and later confirmed through administering
two doses of pyridoxine (he did not react to the first one) and DNA analysis later.
The genetic analysis revealed mutations in both alleles of thegene responsible for encoding Antiquitin, a protein involved in lysine metabolism. Neither of us, Andre’s parents, exhibited symptoms of Pyridoxine-Dependent Epilepsy (PDE) because our healthy alleles compensated for the mutated ones in our son. Andre’s mutations resulted in a non-functional protein and an abnormal amino acid substitution, contributing to his condition. Andre now receives daily pyridoxine supplementation, which has kept his seizures at bay, but faces other challenges in development as compared to healthy children.
Despite the challenges, Andre, at almost 3 years old, is a remarkable child, understanding
commands in both Russian and English. Although he has not started speaking, our hope
remains strong.
We remain grateful for the diagnosis by Dr. Lerner, but we mourn the critical time lost due to the
initial misdiagnosis. Our story is a testament to the importance of early detection and competent
medical care in the face of rare diseases like Pyridoxine-Dependent Epilepsy (PDE). We share
our experience with the hope that it may guide others on their own journeys and contribute to
greater awareness of this condition.


